DeeDeeExperiment is an S4 class extending the SingleCellExperiment framework to facilitate the integration and management of omics analysis results. It introduces two dedicated slots to store Differential Expression analysis (DEA) results and Functional Enrichment analysis (FEA) results, providing a structured approach for downstream analysis.
Installation
You can install the development version of DeeDeeExperiment from GitHub with
library("remotes")
remotes::install_github("imbeimainz/DeeDeeExperiment",
dependencies = TRUE,
build_vignettes = TRUE)Structure and Usage
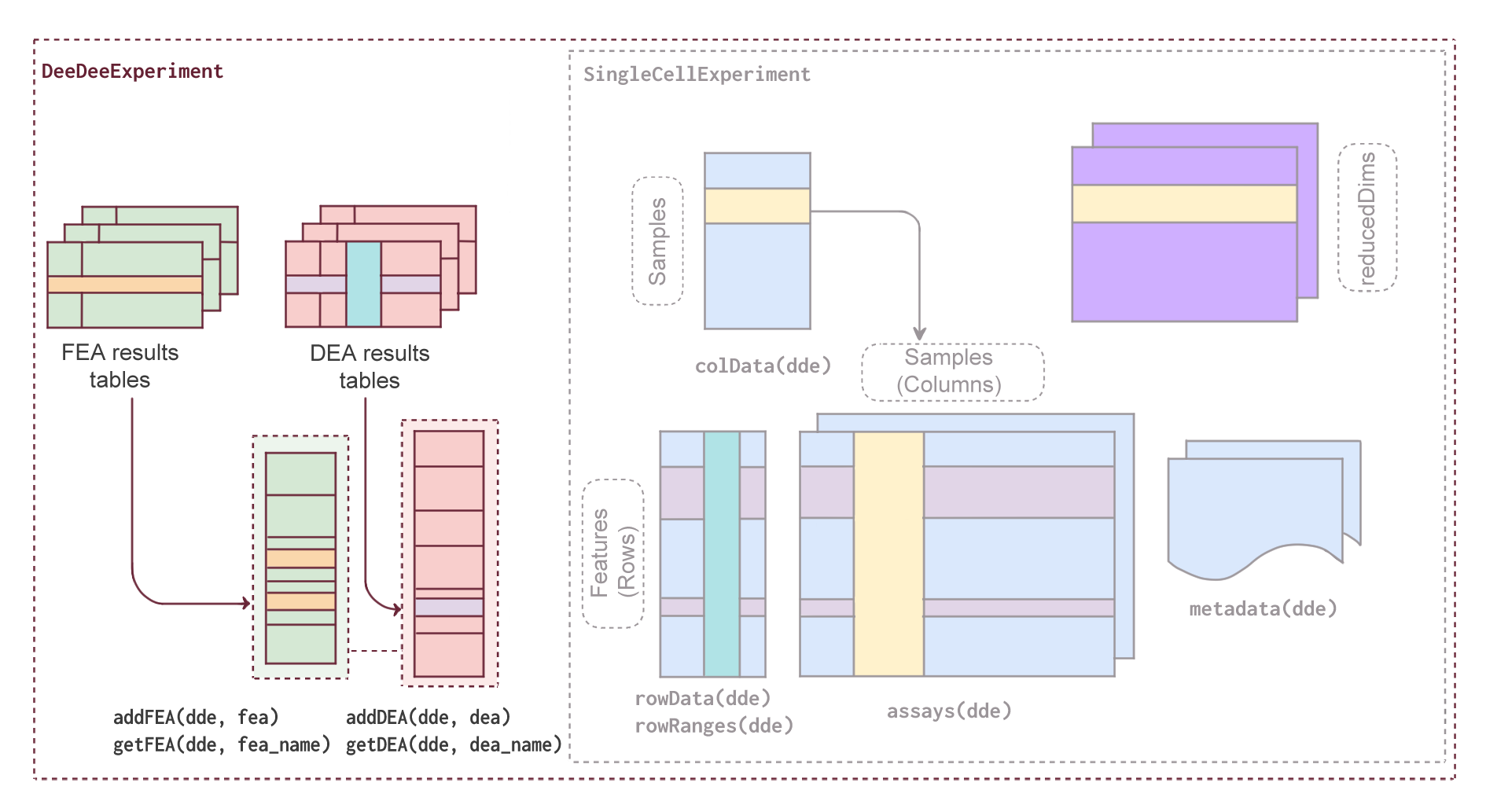
The DeeDeeExperiment class extends the core Bioconductor SingleCellExperiment object, retaining its structure, methods, and compatibility with existing tools. In addition, it introduces new components designed to simplify and enhance downstream analysis.
Specifically, DeeDeeExperiment has two new slots:
dea: A slot that stores results from differential expression analysis (DEA), along with relevant metadata (currently supports results fromDESeq2,edgeR,limma,muscat)fea: A slot that stores results from functional enrichment analysis (FEA), along with relevant metadata (currently supports results fromtopGO,clusterProfiler,enrichR,gProfiler,fgsea,gsea,DAVID, and output ofGeneTonicshakers)
Example
library("DeeDeeExperiment")
library("macrophage")
# load data
data(gse, "macrophage")
data("de_named_list", package = "DeeDeeExperiment")
data("topGO_results_list", package = "DeeDeeExperiment")
dds_macrophage <- DESeq2::DESeqDataSet(gse, design = ~ line + condition)
# create DeeDeeExperiment object
dde <- DeeDeeExperiment(sce = dds_macrophage,
de_results = de_named_list,
enrich_results = topGO_results_list)Development
If you encounter a bug, have usage questions, or want to share ideas and functionality to make this package better, feel free to file an issue.